BEAST, Bayesian evolutionary analysis sampling trees
Alexei Drummond, Bioinformatics, Department of Computer Science
BEAST is a cross-platform program for Bayesian MCMC analysis of molecular sequences. It is entirely oriented towards rooted, time- measured phylogenies inferred using strict or relaxed molecular clock models. It can be used as a method of reconstructing phylogenies but is also a framework for testing evolutionary hypotheses without conditioning on a single tree topology. BEAST uses MCMC to explore tree space in such a way that each tree is weighted proportional to its posterior probability. We include a simple to use user-interface program for setting up standard analyses and a suit of programs for analysing the results.
BEAST 2 (www.beast2.org) develops new features to allow resuming an MCMC chain, and real time tracking of ESSs while running a chain.
It is easily extendible, for example, the BEAST 2 packages supports multi-chain MCMC, some experimental likelihood calculations that are potentially faster than the base implementation, and a spread sheet GUI for manipulating models.
BEAST 2 has a plug-in facility. BEAST 2 packages provide more models, such as
- Birth Death Serial Skyline Model
- Reversible jump substitution model
- SNAPP Species trees from SNP and AFLP analysis
- Substitution Bayesian Model Averaging
- Sampled Ancestors
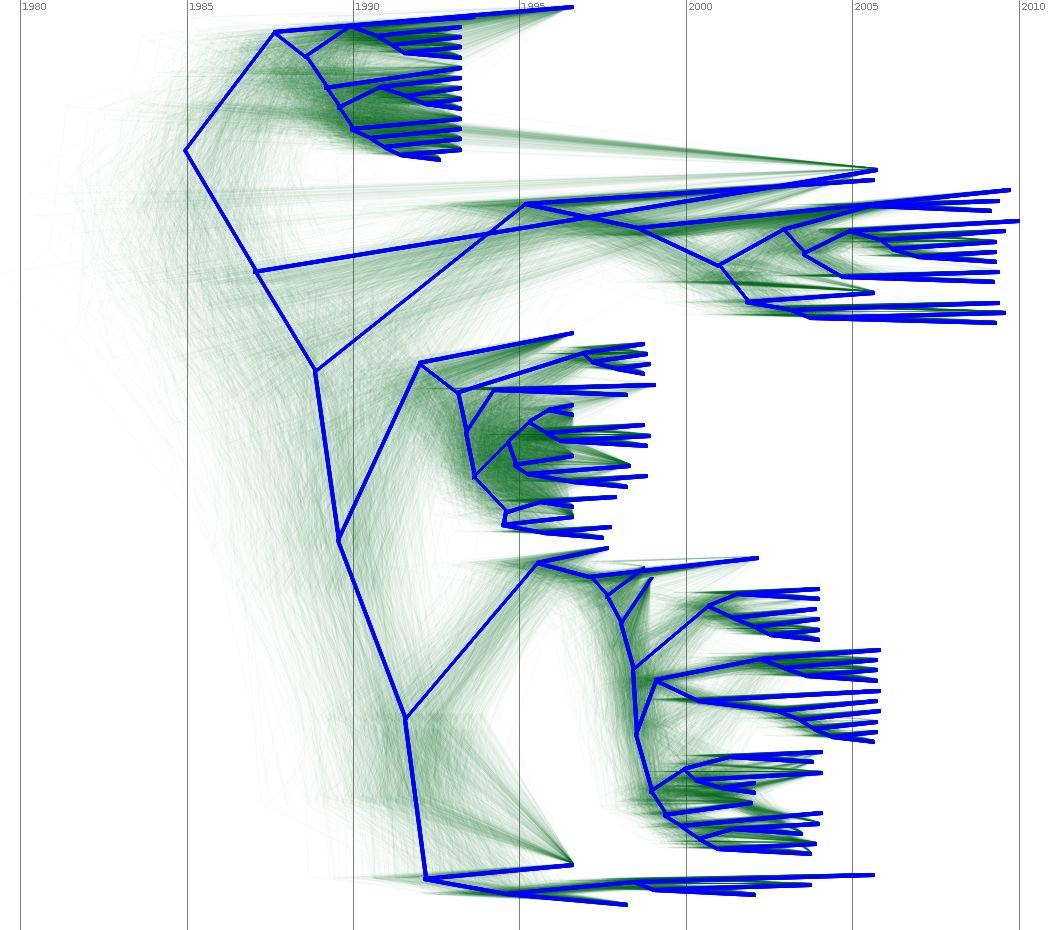
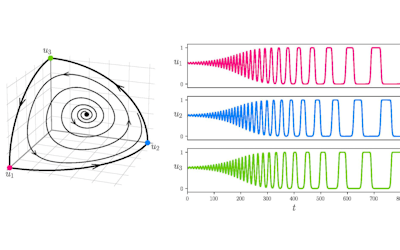
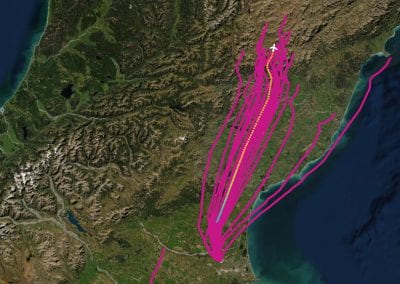
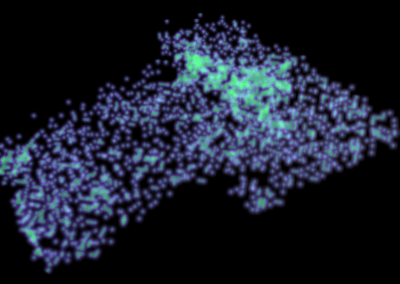
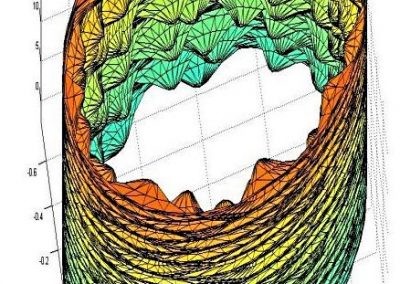
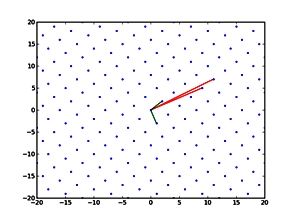
Output of a BEAST analysis of viral samples taken over different years — samples from the same year tend to cluster together. Blue line represents a summary tree of the posterior sample. Green lines indicate uncertainty in the tree distribution. It illustrates how both topology and node height estimates vary in the posterior sample.
They can be easily installed/un-installed from BEAUti (BEAST’s GUI). More packages are being developed (http://www.beast2.org/beagle-beast-2-in-cluster) .
BEAST developers use the NeSI Pan cluster for running simulation studies to validate new models, and run large analyses with questions of considerable scientific interest. For example, BEAST was used for dating the split of Cichlid species between Africa and South America, determining the origin of Indo-European languages (see http://language.cs.auckland.ac.nz), and analysing epidemiological histories of diseases such as HIV and influenza.