High-resolution cryo-electron microscopy of protein complexes and machines
Dr Ambroise Desfosses, Research Fellow, Hari Venugopal, Teaching and Research Technician, Assoc Prof Alok K. Mitra, Department of Biological Sciences
Overview
In all walks of life, functioning of a living system is driven by proteins, the “workhorses” that direct the replication and survival of the constituting biological cell. Be it in a single cell or multi-cellular organism, the essential physiological processes require the interplay of thousands of protein molecules, each assuming an unique, folded 3-D structure, and each executing its biological function efficiently and with great specificity. In recent years, as part of Structural Biology, cryo-electron microscopy has developed into a powerful discipline. This method allows one to reveal high-resolution details of protein structure and protein-protein interaction in macromolecuar complexes to provide insight into many cellular functions that are important in human health and disease.
As part of our program to study the 3-D structures of complex protein assemblies, we use advanced image processing algorithms to generate 3-D structures from images recorded in our state-of-the-art 200kV transmission electron microscope equipped with an extremely coherent field emission gun as the electron source (TF20). These algorithms are used to analyze 2-D projection images of the 3-D complex, whereupon the knowledge of the orientation of the 3-D object that gave rise to individual 2-D image is deduced iteratively from an initial, approximate, reference model of the 3-D structure. This knowledge is then utilised to arrive at the 3-D structure from the many thousands of aligned images. This, so-called single-particle-analysis (SPA) for solving the 3-D structure is computationally demanding especially for large images containing near atomic resolution details.
Utility of the NeSI Pan cluster
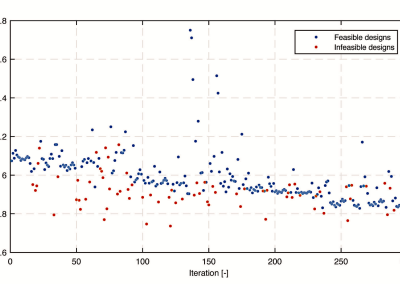
We benefited from the ability to install parallelized versions of modern SPA softwares namely, Bsoft, EMAN2, SPIDER, Relion and most recently SPRING on the NeSI Pan High Performance Computing cluster, which provided exceptional computational power to fully utilise the power of these tools. Obtaining high-resolution information on a structure of interest depends mostly on the amount of 2D images combined in a single 3D structure, the fine sampling of each image, and a precise orientation determination. With a pixel corresponding to only 1 Angstrom (= 10-10 m) at the specimen level, each of the several ten thousands particle image gets as big as 500*500 pixels. Those images need to be compared to ten thousands of projections of a reference volume to determine the optimal translation and rotation to match the reference image. Therefore, in total, billions of cross-correlation have to be computed, and this for each iteration of the refinement. On a typical 4 CPUs machine, a full refinement would take up to a year ! Thanks to the large number of CPU’s available on the NeSI Pan cluster, we can bring down those calculation to less than a week, allowing us also to test and optimise a number of crucial parameters for the 3-dimensional structure determination.
Below, we provide details focusing on the progress in our cryo-EM derived 3-D structural analysis of two macromolecular complexes at the highest resolution.
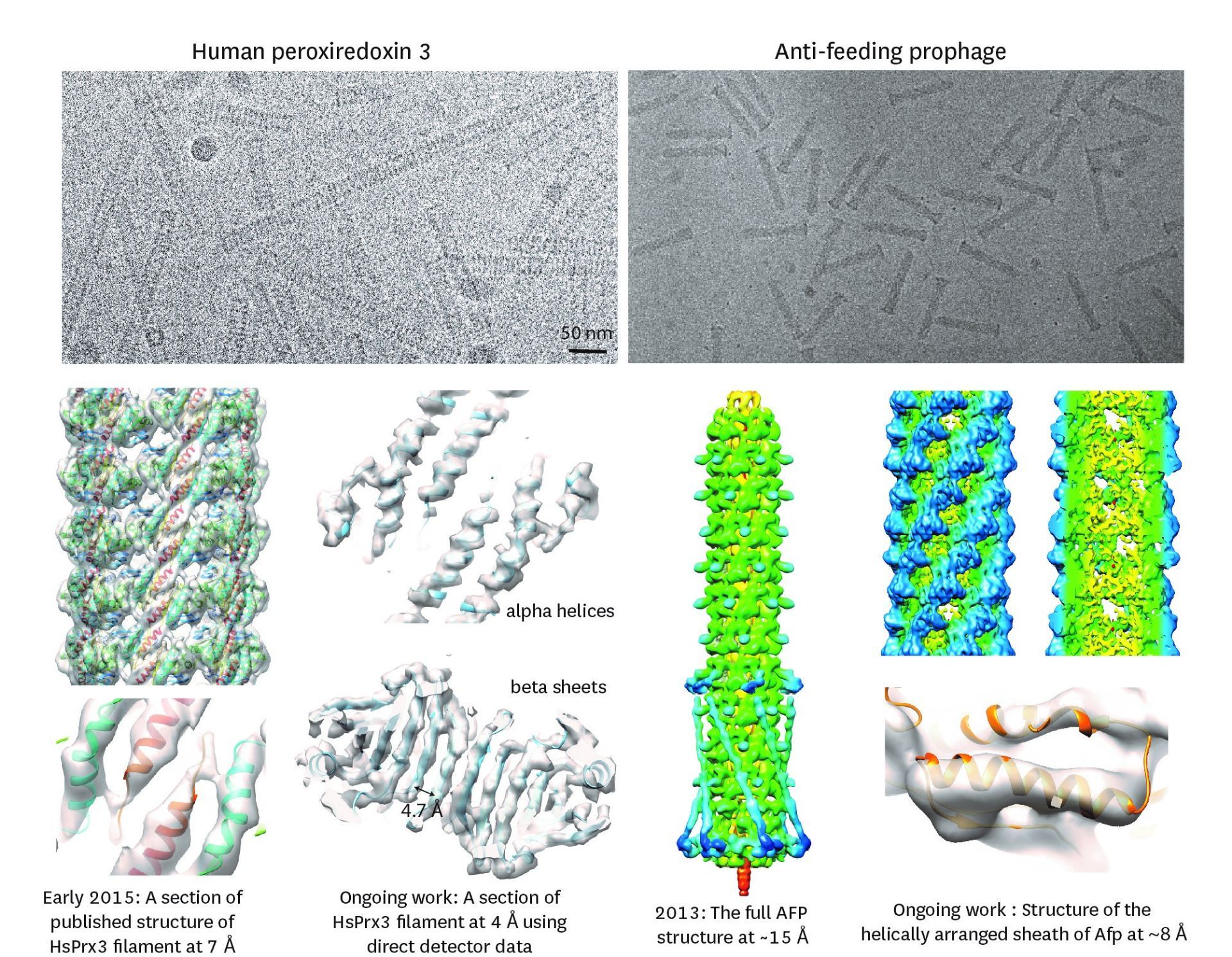
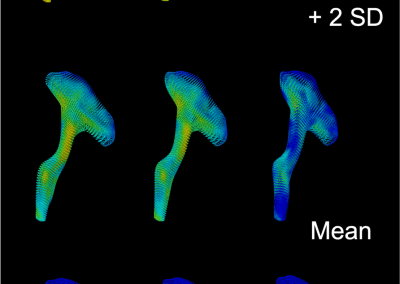
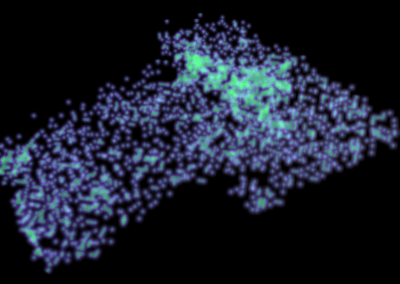
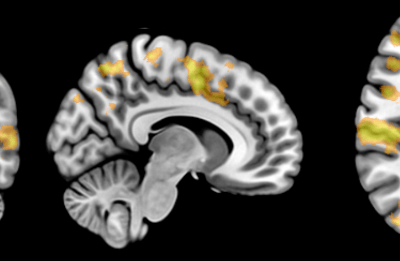
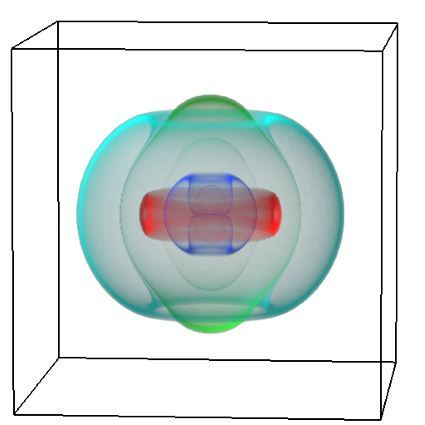
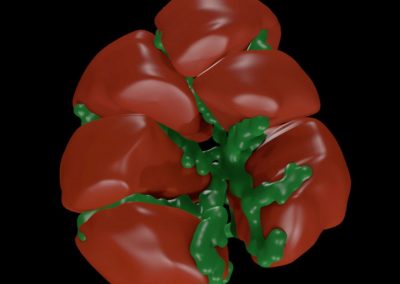
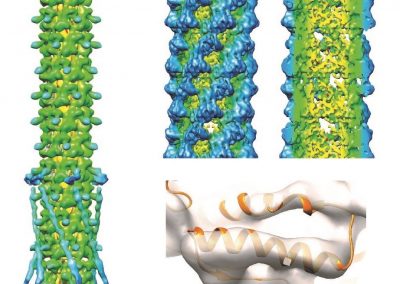
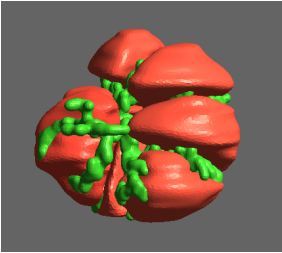
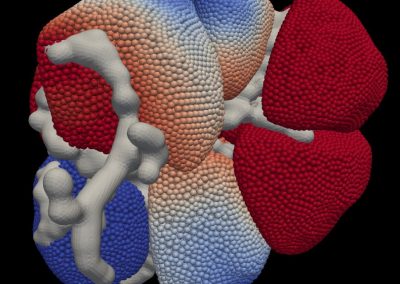
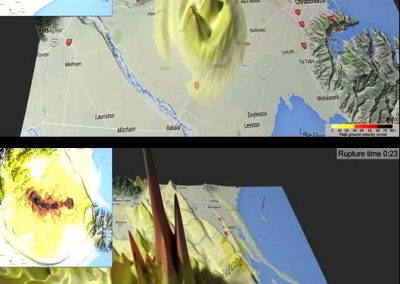
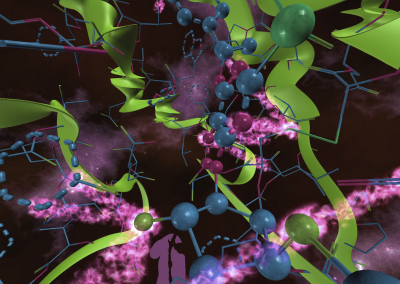
Figure 1: Top, Cryo-EM Micrographs of Human Peroxiredoxin 3 (left) and Anti-feeding prophage (right). From hundreds of such micrographs, the particles are segmented into tens of thoousands of images for which the relative orientations need to be determined. Bottom, example reconstructions obtained for both projects using the NeSI Pan cluster. Three-dimensional structures of those large protein complexes constitute a critical step towards the understanding of their biological function.
Insight into the function of a toxin-delivering protein nano machine
Many strains of bacteria have developed sophisticated protein assemblages that they use to gain a survival niche by either killing other sensitive bacteria through perforation of their membranes or by transferring toxins into eukaryotic hosts. Anti-feeding prophage (Afp) is a toxin-delivering micro-injection apparition released by gram-negative bacteria S. entomophila that is pathogenic to New Zealand agriculture pest grass grub (C. zealandica). Due to the fact that unlike for many other related systems Afp has a eukaryotic host, this has raised the possibility that apart from the benefit of using it as a chemical-free biological pesticide, Afp may be tailored for targeted delivery in anti-tumour immunotherapy. In order to facilitate such exercises, it is mandatory to reveal its detailed 3-D structure, which is composed of no less than 18 different proteins. Afp elaborates two different configurations – the resting extended state and the functional contracted state that is believed to enable extrusion of the toxin into the host (Fig. 1). We have in 2013 produced a modest (~15Å) resolution structure of the extended form by SPA coupled with specialised analysis of the helical symmetry of the sheath (1). Currently, using images from our TF20 microscope we are utilizing the Pan cluster to process simultaneously close to 10,000 images to arrive at a sub nanometre resolution 3-D structure for Afp as also for a structural mutant, the tube-baseplate complex (TBC) for an in-depth visualisation of the inner tube and the baseplate. This work is in collaboration with Dr. Mark Hurst of AgResearch Lincoln.
Insight into the 3-D architecture of a putative molecular chaperone
This study involved revealing the organization of high-molecular weight filamentous assembly of human peroxiredoxin3 (HsprX3). Peroxiredoxins (Prxs) are a ubiquitous class of thiol (Cys)-dependent peroxidases that play an important role in the protection and response of cells to oxidative stress and therefore is clinically very important. The catalytic unit of typical 2-Cys prx is a homodimer, which can self-associate to form complex assemblies that are hypothesized to have signaling and chaperone activity. Mitochondrial Prx3 forms dodecameric toroids, which can further stack to form filaments, the so-called high-molecular-weight (HMW) form that has putative chaperone activity. We imaged such structures formed at low pH using our TF20. We established the helical nature of these filaments and image processing using this deduced helical symmetry resulted in a ~7Å reconstruction (2), the highest resolution cryo-EM structure (Fig. 1) obtained using in-house data in whole of Australasia. We are currently pushing the resolution of this structure by processing data acquired on an electron microscope equipped with a direct detector. This structure establishes the molecular insight into the formation of the helical stacks of PrX3 dodecamers, and provides an explanation of the chaperone activity based on the elaboration of significant hydrophobic patches in the lumen of the filament. This work is in collaboration with Dr. Juliet Gerrard at SBS.
References
- Heyman et al. Three-dimensional structure of the toxin-delivery particle antifeeding prophage of Serratia entomophila. J Biol Chem. 2013
- Radjainia et al. Cryo-Electron Microscopy Structure of Human Peroxiredoxin-3 Filament Reveals the Assembly of a Putative Chaperone. Structure 2015.