Finite element method code for modelling biological cells
Dr John Rugis, Department of Mathematics, Dr Chris Scott, NeSI, Professor James Sneyd, Department of Mathematics, University of Auckland
Background
A collaborative consultation project, aimed at optimising the performance of a custom finite element method (FEM) code, has been undertaken. The aim of this work was to enable the researchers to scale their simulations up and make the best use of NeSI’s High Performance Computing (HPC) platforms. The primary goals were to obtain the best possible performance from the code, while ensuring its robustness and ease of use. The main developer of the code was John Rugis, while support for this project was provided by Chris Scott, through NeSI’s Scientific Programming Consultancy Service.
The research
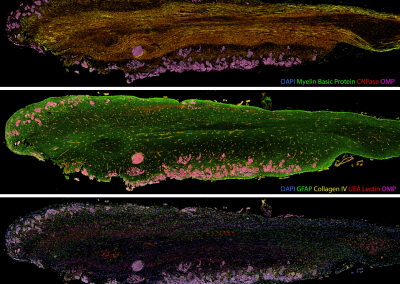
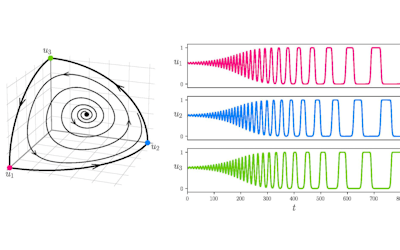
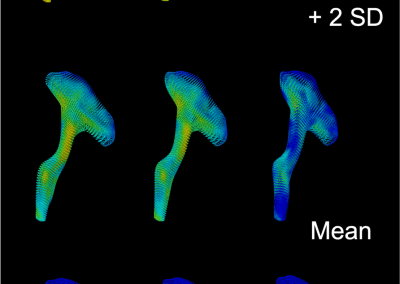
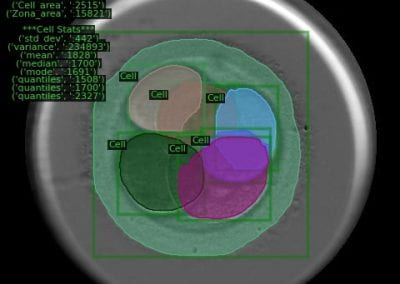
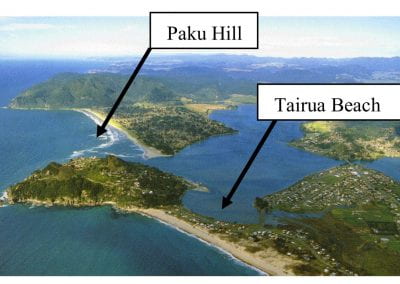
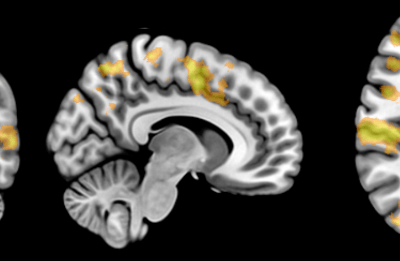
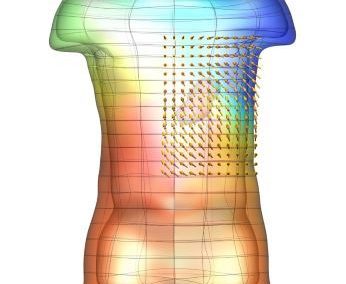
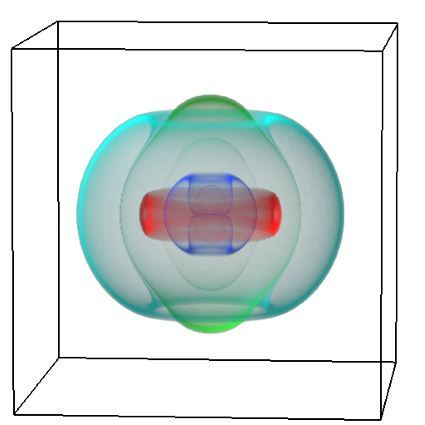
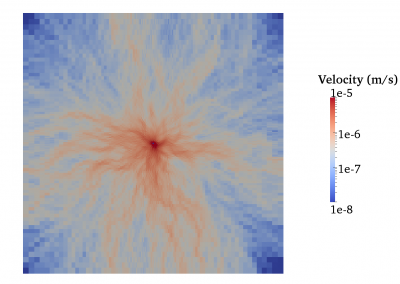
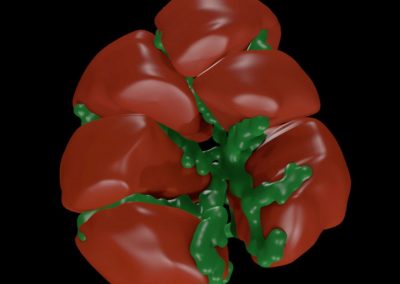
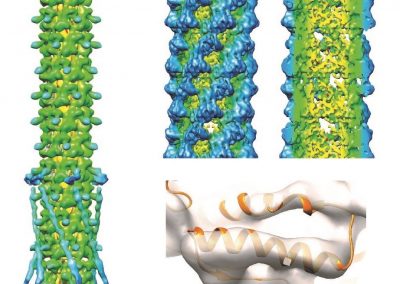
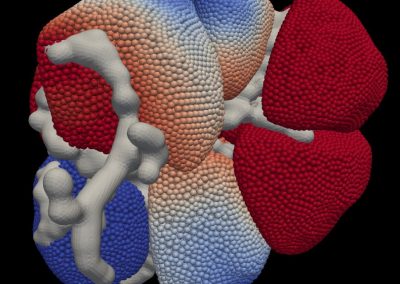
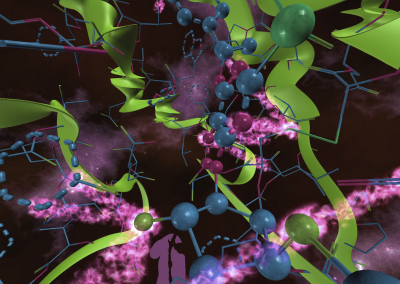
The FEM code is used to model salivary gland acinar cells (shown in Figure 1) by Professor James Sneyd and his research group (Department of Mathematics, University of Auckland). Understanding how salivary glands work to secrete saliva has important applications for oral health, since a lack of saliva can cause a range of medical conditions, such as dry mouth (xerostomia). With the aid of NeSI’s HPC facility the research group have been using mathematical modelling to answer this interesting physiological question.
As part of this project a series of benchmarks and comparisons were performed between libraries for solving sparse linear systems, such as those arising from the FEM method. The ViennaCL library was eventually selected, as it was found to perform very well and supports a variety of parallelisation options (CUDA, OpenCL and OpenMP), allowing the use of accelerators such as GPUs.
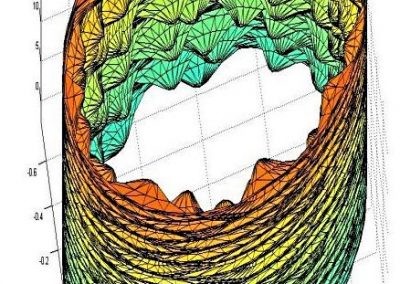
Profiling your code is an important step to ensure there are no unexpected bottlenecks. When profiling the serial version of this code it was found that only a small proportion of the run time was spent solving the sparse linear system, with a large amount of time spent doing other operations outside of the solver. After further investigation, an 8x increase in performance was gained by changing the matrix storage type and thus removing this bottleneck. After making these improvements we benchmarked the different versions of the program, with the results displayed in Figure 2, showing that the GPU version performed twice as fast as the serial version.
Maintaining a single Makefile for the code was cumbersome, since there were a number of different options that had to be chosen at compile time and different rules were required for each machine, which resulted in frequent editing of the Makefile. To alleviate these issues the Makefile was replaced with the CMake build system generator, allowing the options to be selected using command line arguments to CMake. Other nice features of CMake include out-of-source builds, allowing multiple versions to be compiled at once, and the ability to automatically locate dependencies.
Continuous Integration (CI) testing was implemented using the Travis-CI service, which is easy to integrate with GitHub and free for open source projects. With CI, every time a commit is pushed to the repository, or a pull request created, the code is compiled and the tests are run. This means we find out immediately if a change has broken the code and works well when multiple developers are collaborating. A weekly test of the code on the NeSI Pan cluster was also implemented. This testing gives the researchers confidence that their simulations are behaving as expected.
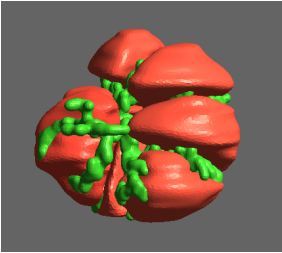
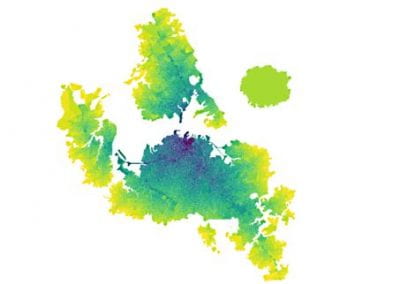
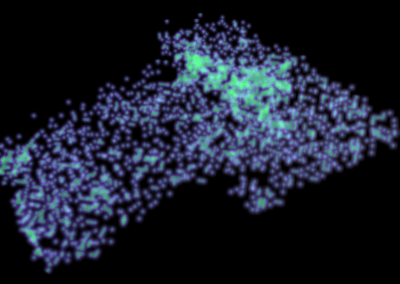
Figure 1. Full 3D model of a cluster of parotid acinar cells.
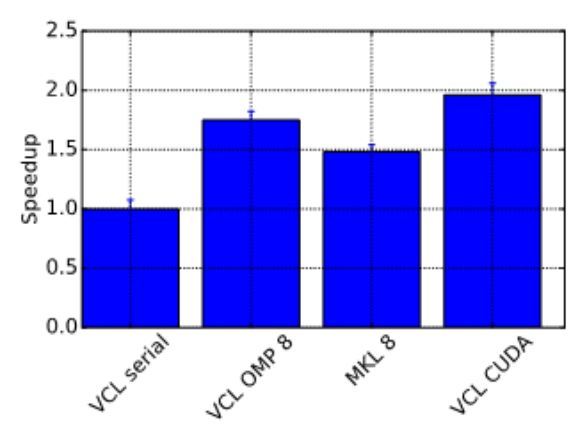
Figure 2. Benchmark results showing the speedup obtained from the ViennaCL OpenMP (8 cores) and CUDA and MKL (8 cores) versions.
This code is typically used to perform parameter sweeps, which can involve large numbers of simulations. For example, varying 2 parameters, with each taking 5 values, for the 7 individual cells in the model, results in 175 simulations. To facilitate the submission of these types of jobs a runtime environment was created, where there is a single config file to be edited, defining the sweep, and a single Python script that launches the set of simulations. Another Python script can be used to generate graphs from the simulations, making it easy to visualise the results.
As a result of this project the researcher’s simulations are now running faster, it is easier for the researchers to submit and analyse large parameter sweeps and robust software development practices are being utilised, giving the researchers confidence in their results and making future development easier. Recent results from this work (see DOI:10.1016/j.jtbi.2016.04.030) have investigated calcium dynamics in individual cells. The next stages of the project are to incorporate fluid flow within the model and also to couple individual cells together to model the full cluster of parotid acinar cells.
Links
https://github.com/jrugis/cellsim_34_vcl (repository for the FEM code)
https://github.com/jrugis/CellSims (repository for the runtime environment)
https://travis-ci.org/jrugis/cellsim_34_vcl (Travis-CI builds page for the FEM code)